Streamlined Data Summary in R STUDIO: Enhancing Bar Graphs with Error Bars
When working with data in R, there are situations where you might need to examine summarized information, such as means, standard deviations, and more. Today, I will introduce the methods that can be employed for this purpose.
Let’s start by loading a dataset.
install.packages("readr")
library(readr)
github= "https://raw.githubusercontent.com/agronomy4future/raw_data_practice/main/fertilizer_treatment.csv"
dataA= data.frame(read_csv(url(github),show_col_types = FALSE))
Genotype Block variable value
1 Genotype_A I Control 42.9
2 Genotype_A II Control 41.6
3 Genotype_A III Control 28.9
4 Genotype_A IV Control 30.8
5 Genotype_B I Control 53.3
6 Genotype_B II Control 69.6
7 Genotype_B III Control 45.4
8 Genotype_B IV Control 35.1
9 Genotype_C I Control 62.3
10 Genotype_C II Control 58.5
11 Genotype_C III Control 44.6
12 Genotype_C IV Control 50.3
13 Genotype_D I Control 75.4
14 Genotype_D II Control 65.6
15 Genotype_D III Control 54.0
16 Genotype_D IV Control 52.7
17 Genotype_A I Fertilizer1 53.8
18 Genotype_A II Fertilizer1 58.5
19 Genotype_A III Fertilizer1 43.9
20 Genotype_A IV Fertilizer1 46.3
21 Genotype_B I Fertilizer1 57.6
22 Genotype_B II Fertilizer1 69.6
23 Genotype_B III Fertilizer1 42.4
24 Genotype_B IV Fertilizer1 51.9
25 Genotype_C I Fertilizer1 63.4
26 Genotype_C II Fertilizer1 50.4
27 Genotype_C III Fertilizer1 45.0
28 Genotype_C IV Fertilizer1 46.7
29 Genotype_D I Fertilizer1 70.3
30 Genotype_D II Fertilizer1 67.3
31 Genotype_D III Fertilizer1 57.6
32 Genotype_D IV Fertilizer1 58.5
33 Genotype_A I Fertilizer2 49.5
34 Genotype_A II Fertilizer2 53.8
35 Genotype_A III Fertilizer2 40.7
36 Genotype_A IV Fertilizer2 39.4
37 Genotype_B I Fertilizer2 59.8
38 Genotype_B II Fertilizer2 65.8
39 Genotype_B III Fertilizer2 41.4
40 Genotype_B IV Fertilizer2 45.4
41 Genotype_C I Fertilizer2 64.5
42 Genotype_C II Fertilizer2 46.1
43 Genotype_C III Fertilizer2 62.6
44 Genotype_C IV Fertilizer2 50.3
45 Genotype_D I Fertilizer2 68.8
46 Genotype_D II Fertilizer2 65.3
47 Genotype_D III Fertilizer2 45.6
48 Genotype_D IV Fertilizer2 51.0
49 Genotype_A I Fertilizer3 44.4
50 Genotype_A II Fertilizer3 41.8
51 Genotype_A III Fertilizer3 28.3
52 Genotype_A IV Fertilizer3 34.7
53 Genotype_B I Fertilizer3 64.1
54 Genotype_B II Fertilizer3 57.4
55 Genotype_B III Fertilizer3 44.1
56 Genotype_B IV Fertilizer3 51.6
57 Genotype_C I Fertilizer3 63.6
58 Genotype_C II Fertilizer3 56.1
59 Genotype_C III Fertilizer3 52.7
60 Genotype_C IV Fertilizer3 51.8
61 Genotype_D I Fertilizer3 71.6
62 Genotype_D II Fertilizer3 69.4
63 Genotype_D III Fertilizer3 56.6
64 Genotype_D IV Fertilizer3 47.4As I engage in various tasks involving this data, I aim to summarize it. Therefore, I will introduce methods applicable to such situations.
1) using plyr package
First, install and activate the package.
install.packages ("plyr")
library (plyr)I want to summarize the average values for the ‘Genotype’ and ‘variable’ in the given dataset “dataA”. The summarized data will be named “dataB”.
dataB= ddply (dataA, c("Genotype","variable"), summarise, mean=mean(value), sd=sd(value), n=length(value), se=sd/sqrt(n))
Genotype variable mean sd n se
1 Genotype_A Control 36.050 7.220572 4 3.610286
2 Genotype_A Fertilizer1 50.625 6.733684 4 3.366842
3 Genotype_A Fertilizer2 45.850 6.943822 4 3.471911
4 Genotype_A Fertilizer3 37.300 7.266820 4 3.633410
5 Genotype_B Control 50.850 14.552548 4 7.276274
6 Genotype_B Fertilizer1 55.375 11.368487 4 5.684244
7 Genotype_B Fertilizer2 53.100 11.581019 4 5.790509
8 Genotype_B Fertilizer3 54.300 8.504509 4 4.252254
9 Genotype_C Control 53.925 7.982637 4 3.991319
10 Genotype_C Fertilizer1 51.375 8.327615 4 4.163807
11 Genotype_C Fertilizer2 55.875 9.059939 4 4.529970
12 Genotype_C Fertilizer3 56.050 5.363146 4 2.681573
13 Genotype_D Control 61.925 10.692482 4 5.346241
14 Genotype_D Fertilizer1 63.425 6.336863 4 3.168432
15 Genotype_D Fertilizer2 57.675 11.139532 4 5.569766
16 Genotype_D Fertilizer3 61.250 11.357670 4 5.678835The means, standard deviations (sd), and standard errors (se) of the values are compiled for each combination of Genotype and variable.
□ Utilizing R Studio for Data Grouping and Mean/Standard Error Calculation (feat ddply)
2) using dplyr package
This time, I will demonstrate how to create the same data using the dplyr package. First, let’s install the package.
install.packages ("dplyr")
library (dplyr)I will create a dataset that compiles the means, standard deviations, and standard errors of value based on Genotype and variable in the given dataset “dataA”. The summarized data will be named “dataC”.
The %>% symbol can be generated automatically by pressing Ctrl + Shift + M, which eliminates the requirement for manual typing.
dataC= dataA %>%
group_by(Genotype, variable) %>%
summarise(mean=mean(value), sd=sd(value), n=length(value), se=sd/sqrt(n))
# A tibble: 16 × 6
# Groups: Genotype [4]
Genotype variable mean sd n se
<chr> <chr> <dbl> <dbl> <int> <dbl>
1 Genotype_A Control 36.0 7.22 4 3.61
2 Genotype_A Fertilizer1 50.6 6.73 4 3.37
3 Genotype_A Fertilizer2 45.8 6.94 4 3.47
4 Genotype_A Fertilizer3 37.3 7.27 4 3.63
5 Genotype_B Control 50.8 14.6 4 7.28
6 Genotype_B Fertilizer1 55.4 11.4 4 5.68
7 Genotype_B Fertilizer2 53.1 11.6 4 5.79
8 Genotype_B Fertilizer3 54.3 8.50 4 4.25
9 Genotype_C Control 53.9 7.98 4 3.99
10 Genotype_C Fertilizer1 51.4 8.33 4 4.16
11 Genotype_C Fertilizer2 55.9 9.06 4 4.53
12 Genotype_C Fertilizer3 56.0 5.36 4 2.68
13 Genotype_D Control 61.9 10.7 4 5.35
14 Genotype_D Fertilizer1 63.4 6.34 4 3.17
15 Genotype_D Fertilizer2 57.7 11.1 4 5.57
16 Genotype_D Fertilizer3 61.2 11.4 4 5.68In the provided data, you might notice that it’s labeled as a tibble. This means that the data is in the tibble format, not a regular data frame. In reality, there isn’t a significant difference in terms of data analysis. I also looked it up since I wasn’t sure, and the differences are well explained on the webpage below:
https://sulgik.github.io/r4ds/tibble.html
If you want to convert the tibble format to a data frame format, you can do so using the following approach
dataC= dataA %>%
group_by(Genotype, variable) %>%
summarise(mean=mean(value), sd=sd(value), n=length(value), se=sd/sqrt(n)) %>%
as.data.frame()
Genotype variable mean sd n se
1 Genotype_A Control 36.050 7.220572 4 3.610286
2 Genotype_A Fertilizer1 50.625 6.733684 4 3.366842
3 Genotype_A Fertilizer2 45.850 6.943822 4 3.471911
4 Genotype_A Fertilizer3 37.300 7.266820 4 3.633410
5 Genotype_B Control 50.850 14.552548 4 7.276274
6 Genotype_B Fertilizer1 55.375 11.368487 4 5.684244
7 Genotype_B Fertilizer2 53.100 11.581019 4 5.790509
8 Genotype_B Fertilizer3 54.300 8.504509 4 4.252254
9 Genotype_C Control 53.925 7.982637 4 3.991319
10 Genotype_C Fertilizer1 51.375 8.327615 4 4.163807
11 Genotype_C Fertilizer2 55.875 9.059939 4 4.529970
12 Genotype_C Fertilizer3 56.050 5.363146 4 2.681573
13 Genotype_D Control 61.925 10.692482 4 5.346241
14 Genotype_D Fertilizer1 63.425 6.336863 4 3.168432
15 Genotype_D Fertilizer2 57.675 11.139532 4 5.569766
16 Genotype_D Fertilizer3 61.250 11.357670 4 5.678835
No matter which package is used, the data has been summarized by means. Now, let’s proceed to create a bar graph using this summarized mean data.
library(ggplot2)
ggplot(data=dataB, aes(x=Genotype, y=mean, fill=variable))+
geom_bar(stat="identity",position="dodge", width = 0.7, size=1) +
geom_errorbar(aes(ymin= mean-se, ymax=mean + se), position=position_dodge(0.7),
width=0.2, color='Black') +
scale_fill_manual(values= c ("dark blue", "darkred", "blue", "orange")) +
scale_y_continuous(breaks = seq(0,100,20), limits = c(0,100)) +
labs(x="Genotype", y="Yield") +
theme_classic(base_size=20, base_family="serif")+
theme(legend.position=c(0.90,0.9),,
legend.title=element_blank(),
legend.key.size=unit(0.5,'cm'),
legend.key=element_rect(color=alpha("white",.05),
fill=alpha("white",.05)),
legend.text=element_text(size=11),
legend.background= element_rect(fill=alpha("white",.05)),
panel.grid.major=element_line(colour="grey90", linewidth=0.5),
axis.line=element_line(linewidth=0.5, colour="black")) +
windows(width=8, height=5) Since you used windows(), the graph will be displayed in a new window.
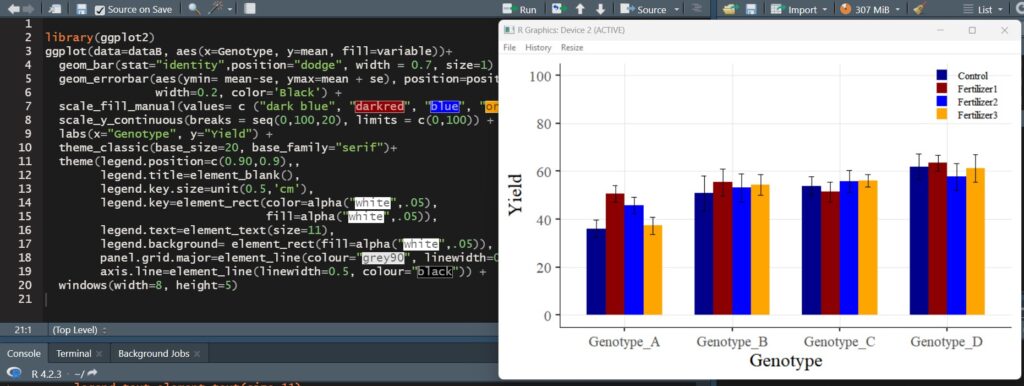
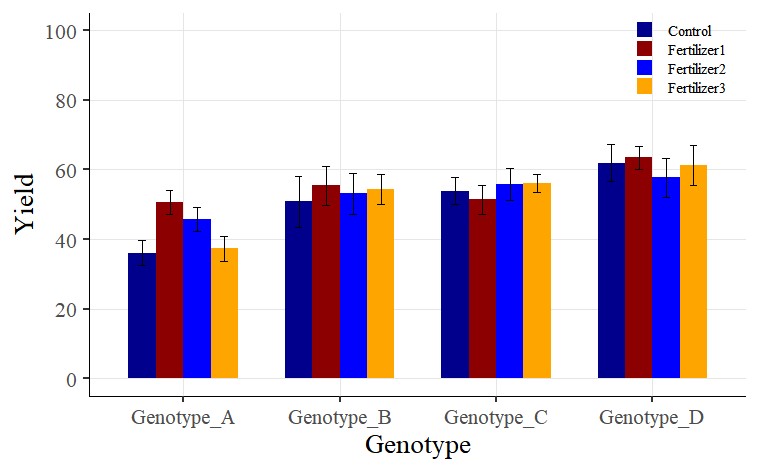
A bar graph with standard errors included has been successfully plotted.
Tip 1 > If you want to change legend titles:
In the above graph, you used the legend.title = element_blank() code to hide the legend title. Now, I want to display the legend title as “Treatment”.
First, I will change the code from legend.title = element_blank() to legend.title = element_text(face= "plain", family= "serif", size= 12, color= "Black"). Additionally, I will include name= "Treatment" in the scale_fill_manual(values= c("dark blue", "darkred", "blue", "orange")) code. In other words, the modified code will be scale_fill_manual(name= "Treatment", values= c("dark blue", "darkred", "blue", "orange")).
The complete code is as follows:
library(ggplot2)
ggplot(data=dataB, aes(x=Genotype, y=mean, fill=variable))+
geom_bar(stat="identity",position="dodge", width = 0.7, size=1) +
geom_errorbar(aes(ymin= mean-se, ymax=mean + se), position=position_dodge(0.7),
width=0.2, color='Black') +
scale_fill_manual(name="Treatment", values= c("dark blue", "darkred", "blue", "orange")) +
scale_y_continuous(breaks = seq(0,100,20), limits = c(0,100)) +
labs (x="Genotype", y="Yield") +
theme_classic(base_size=20, base_family="serif")+
theme(legend.position=c(0.90,0.9),,
legend.title= element_text(face= "plain", family="serif", size= 12, color= "Black"),
legend.key.size=unit(0.5,'cm'),
legend.key=element_rect(color=alpha("white",.05),
fill=alpha("white",.05)),
legend.text=element_text(size=11),
legend.background= element_rect(fill=alpha("white",.05)),
panel.grid.major=element_line(colour="grey90", linewidth=0.5),
axis.line=element_line(linewidth=0.5, colour="black")) +
windows(width=8, height=5)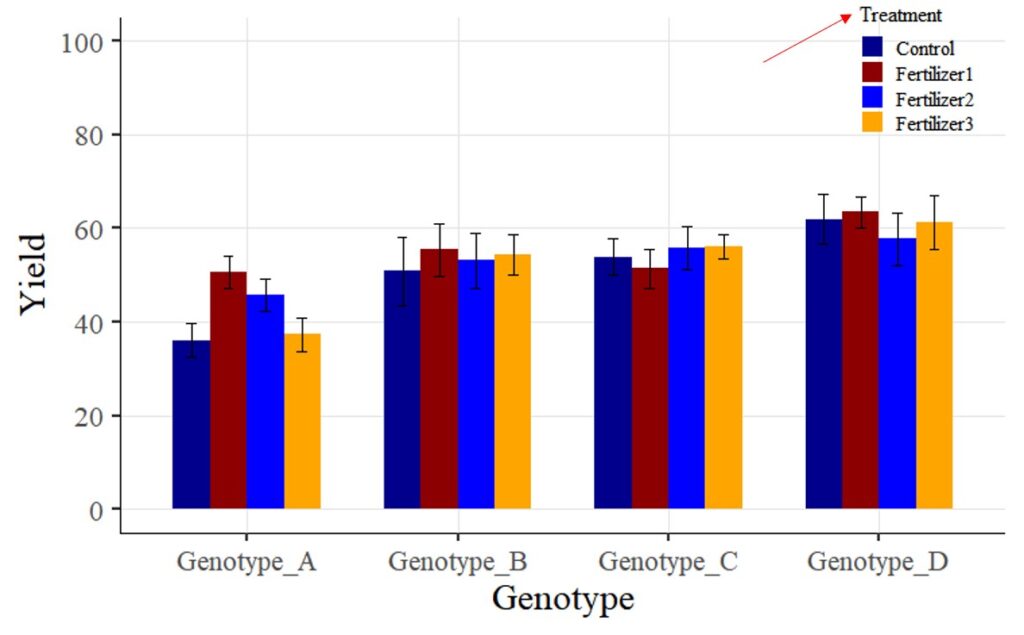
Tip 2 > If you want to change legend labels:
Now, I want to change the legend label ‘Fertilizer 1 – 3’ to ‘Nitrogen,’ ‘Phosphorus,’ and ‘Potassium.’ How can I achieve this? While there are various methods to change variable names, for now, I’ll make the changes directly in the provided codes.
□ How to Rename Variables within Columns in R?
In the scale_fill_manual() function, I will add some code as shown below.
scale_fill_manual(name="Treatment", values= c("dark blue", "darkred", "blue", "orange"),
breaks=c("Control","Fertilizer1","Fertilizer2","Fertilizer3"),
labels=c("Control", "Nitrogen","Phosphorus","Potassium")) +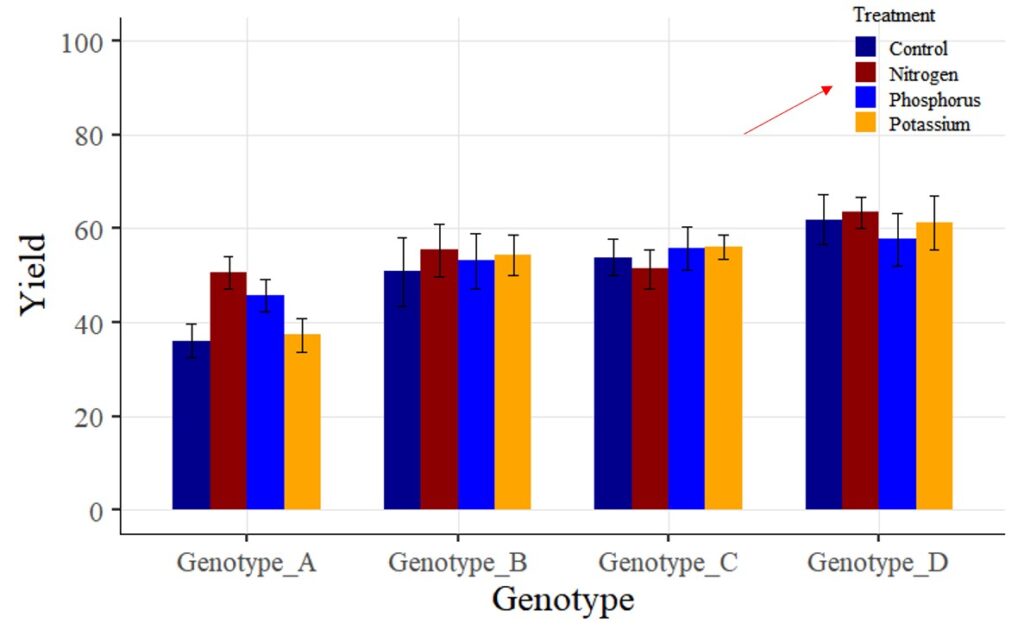
The complete code is provided below.
library(ggplot2)
ggplot(data=dataB, aes(x=Genotype, y=mean, fill=variable))+
geom_bar(stat="identity",position="dodge", width = 0.7, size=1) +
geom_errorbar(aes(ymin= mean-se, ymax=mean + se), position=position_dodge(0.7),
width=0.2, color='Black') +
scale_fill_manual(name="Treatment", values= c("dark blue", "darkred", "blue", "orange"),
breaks=c("Control","Fertilizer1","Fertilizer2","Fertilizer3"),
labels=c("Control", "Nitrogen","Phosphorus","Potassium")) +
scale_y_continuous(breaks = seq(0,100,20), limits = c(0,100)) +
labs (x="Genotype", y="Yield") +
theme_classic(base_size=20, base_family="serif")+
theme(legend.position=c(0.90,0.9),,
legend.title= element_text(face= "plain", family="serif", size= 12, color= "Black"),
legend.key.size=unit(0.5,'cm'),
legend.key=element_rect(color=alpha("white",.05),
fill=alpha("white",.05)),
legend.text=element_text(size=11),
legend.background= element_rect(fill=alpha("white",.05)),
panel.grid.major=element_line(colour="grey90", linewidth=0.5),
axis.line=element_line(linewidth=0.5, colour="black")) +
windows(width=8, height=5)