Combining Factors from Separate Datasets into a Single Column Using R Studio (feat. dplyr package)
When data is divided into two separate datasets, it needs to be combined into a single column. Using R, we can simply combine the two datasets.
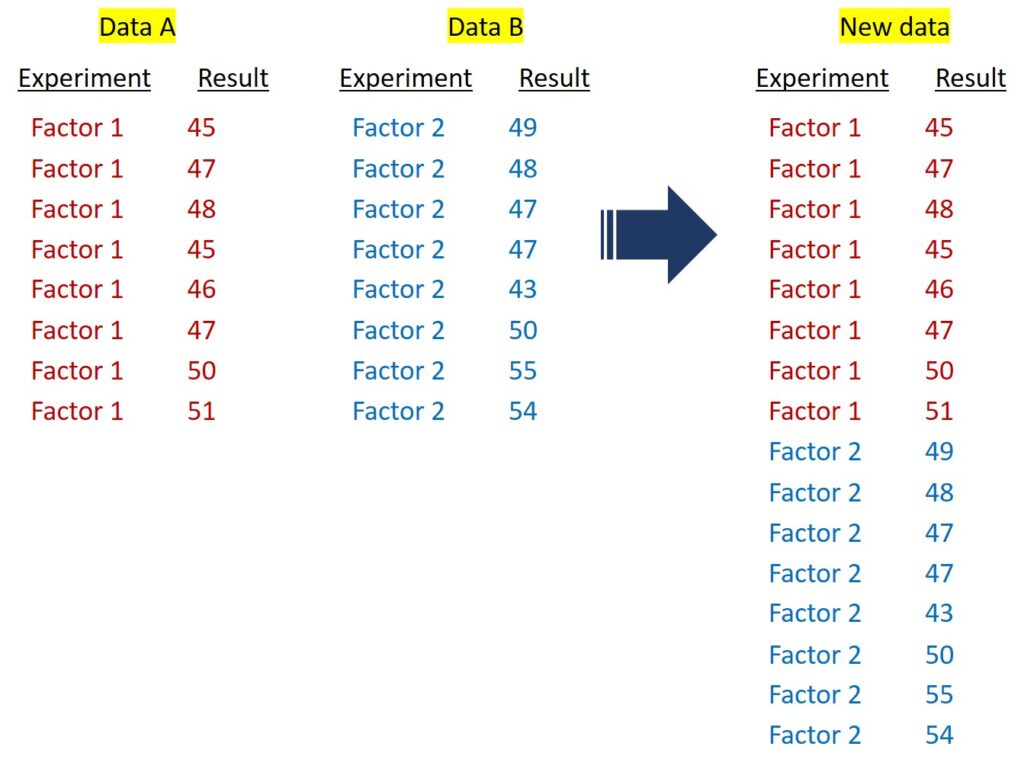
I will create a simple dataset.
Defoliation= rbind(data.frame(Genotype=c ("CV1","CV2","CV3","CV4","CV5"),
Block="I", yield=c("50","52","55","56","54")),
data.frame(Genotype=c ("CV1","CV2","CV3","CV4","CV5"),
Block="II", yield=c("52","50","56","58","59")),
data.frame(Genotype=c ("CV1","CV2","CV3","CV4","CV5"),
Block="III", yield=c("52","50","56","58","59")))
Defoliation
Genotype Block yield
1 CV1 I 50
2 CV2 I 52
3 CV3 I 55
4 CV4 I 56
5 CV5 I 54
6 CV1 II 52
7 CV2 II 50
8 CV3 II 56
9 CV4 II 58
10 CV5 II 59
11 CV1 III 52
12 CV2 III 50
13 CV3 III 56
14 CV4 III 58
15 CV5 III 59
De_graining= rbind(data.frame(Genotype= c("CV1","CV2","CV3","CV4","CV5"),
Block="I", yield= c("62","59","62","60","59")),
data.frame(Genotype= c("CV1","CV2","CV3","CV4","CV5"),
Block="II", yield= c("61","60","63","62","60")),
data.frame(Genotype= c("CV1","CV2","CV3","CV4","CV5"),
Block="III", yield= c("64","58","65","61","62")))
De_graining
Genotype Block yield
1 CV1 I 62
2 CV2 I 59
3 CV3 I 62
4 CV4 I 60
5 CV5 I 59
6 CV1 II 61
7 CV2 II 60
8 CV3 II 63
9 CV4 II 62
10 CV5 II 60
11 CV1 III 64
12 CV2 III 58
13 CV3 III 65
14 CV4 III 61
15 CV5 III 62Now I will combine these two datasets into one.
1) using data.frame()
To explain the below code simply, we are using the function data.frame() to combine two datasets. Regarding the repetition of the text “Defoliation,” it indicates repeating it by the number of times corresponding to the values in the ‘yield’ column of the Defoliation dataset, which is length(Defoliation$yield). Similarly, for the text “De_graining,” it means repeating it by the number of times corresponding to the values in the ‘yield’ column of the De_graining dataset, which is length(De_graining$yield). By doing this repetition, we are taking the values of the ‘yield’ column from the Defoliation dataset and the De_graining dataset.
When you execute the above code, the data will be structured as follows. However, this data lacks an ID. In other words, we need to incorporate the Genotype and Block, which constitute the ID in this data.
new_data= data.frame (dataset=c(rep("Defoliation", length(Defoliation$yield)),
rep("De_graining",length(De_graining$yield))),
value=c(Defoliation$yield, De_graining$yield))
new_data
dataset value
1 Defoliation 50
2 Defoliation 52
3 Defoliation 55
4 Defoliation 56
5 Defoliation 54
6 Defoliation 52
7 Defoliation 50
8 Defoliation 56
9 Defoliation 58
10 Defoliation 59
11 Defoliation 52
12 Defoliation 50
13 Defoliation 56
14 Defoliation 58
15 Defoliation 59
16 De_graining 62
17 De_graining 59
18 De_graining 62
19 De_graining 60
20 De_graining 59
21 De_graining 61
22 De_graining 60
23 De_graining 63
24 De_graining 62
25 De_graining 60
26 De_graining 64
27 De_graining 58
28 De_graining 65
29 De_graining 61
30 De_graining 62So, I will make a slight modification to the code as shown below.
new_data2= data.frame (Defoliation$Genotype, De_graining$Genotype,
Defoliation$Block, De_graining$Block,
dataset=c(rep("Defoliation", length(Defoliation$yield)),
rep("De_graining", length(De_graining$yield))),
value=c(Defoliation$yield,De_graining$yield))As a result, the data structure will be organized as shown below.
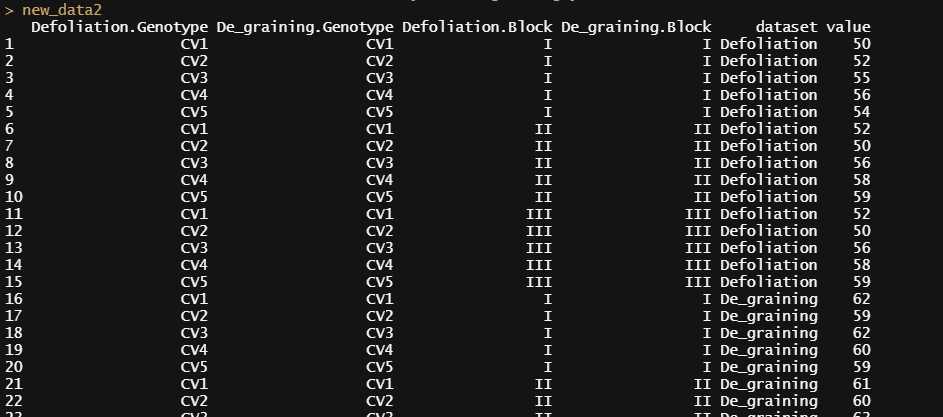
The reason for including Defoliation$Genotype, De_graining$Genotype, Defoliation$Block, and De_graining$Block in the data.frame() function is to verify whether the two datasets have been stacked vertically properly. If both datasets share identical conditions for Genotype and Block, you could include only Defoliation$Genotype and Defoliation$Block. These two columns serve as variable conditions describing the vertically stacked data.
In other words, if you write the code as shown below:
new_data3= data.frame (Defoliation$Genotype, Defoliation$Block,
dataset=c(rep("Defoliation", length(Defoliation$yield)),
rep("De_graining", length(De_graining$yield))),
value=c(Defoliation$yield, De_graining$yield))
new_data3
Defoliation.Genotype Defoliation.Block dataset value
1 CV1 I Defoliation 50
2 CV2 I Defoliation 52
3 CV3 I Defoliation 55
4 CV4 I Defoliation 56
5 CV5 I Defoliation 54
6 CV1 II Defoliation 52
7 CV2 II Defoliation 50
8 CV3 II Defoliation 56
9 CV4 II Defoliation 58
10 CV5 II Defoliation 59
11 CV1 III Defoliation 52
12 CV2 III Defoliation 50
13 CV3 III Defoliation 56
14 CV4 III Defoliation 58
15 CV5 III Defoliation 59
16 CV1 I De_graining 62
17 CV2 I De_graining 59
18 CV3 I De_graining 62
19 CV4 I De_graining 60
20 CV5 I De_graining 59
21 CV1 II De_graining 61
22 CV2 II De_graining 60
23 CV3 II De_graining 63
24 CV4 II De_graining 62
25 CV5 II De_graining 60
26 CV1 III De_graining 64
27 CV2 III De_graining 58
28 CV3 III De_graining 65
29 CV4 III De_graining 61
30 CV5 III De_graining 62The data structure will look as shown above. Let’s change the column names to make the data more clear and understandable.
colnames(new_data3)[1]= c("Genotype")
colnames(new_data3)[2]= c("Block")
colnames(new_data3)[3]= c("Treatment")
colnames(new_data3)[4]= c("Grain weight")
new_data3
Genotype Block Treatment Grain weight
1 CV1 I Defoliation 50
2 CV2 I Defoliation 52
3 CV3 I Defoliation 55
4 CV4 I Defoliation 56
5 CV5 I Defoliation 54
6 CV1 II Defoliation 52
7 CV2 II Defoliation 50
8 CV3 II Defoliation 56
9 CV4 II Defoliation 58
10 CV5 II Defoliation 59
11 CV1 III Defoliation 52
12 CV2 III Defoliation 50
13 CV3 III Defoliation 56
14 CV4 III Defoliation 58
15 CV5 III Defoliation 59
16 CV1 I De_graining 62
17 CV2 I De_graining 59
18 CV3 I De_graining 62
19 CV4 I De_graining 60
20 CV5 I De_graining 59
21 CV1 II De_graining 61
22 CV2 II De_graining 60
23 CV3 II De_graining 63
24 CV4 II De_graining 62
25 CV5 II De_graining 60
26 CV1 III De_graining 64
27 CV2 III De_graining 58
28 CV3 III De_graining 65
29 CV4 III De_graining 61
30 CV5 III De_graining 62The drawback of the data.frame() function is that if the number of rows is different, the datasets cannot be merged. So, in the case above, since the number of rows was the same, they could be combined. This is where the dplyr package comes in as a solution to easily merge data without such issues.
2. using dplyr package
The method mentioned above is quite primitive in terms of R usage. The dplyr package simplifies tasks like the one described above and can accomplish them in a matter of seconds. After installing the package, you can use the bind_rows() function.
install.packages("dplyr")
library(dplyr)
new_data4= bind_rows (Defoliation, De_graining)
new_data4
Genotype Block yield
1 CV1 I 50
2 CV2 I 52
3 CV3 I 55
4 CV4 I 56
5 CV5 I 54
6 CV1 II 52
7 CV2 II 50
8 CV3 II 56
9 CV4 II 58
10 CV5 II 59
11 CV1 III 52
12 CV2 III 50
13 CV3 III 56
14 CV4 III 58
15 CV5 III 59
16 CV1 I 62
17 CV2 I 59
18 CV3 I 62
19 CV4 I 60
20 CV5 I 59
21 CV1 II 61
22 CV2 II 60
23 CV3 II 63
24 CV4 II 62
25 CV5 II 60
26 CV1 III 64
27 CV2 III 58
28 CV3 III 65
29 CV4 III 61
30 CV5 III 62The data has been sorted as columns all at once. However, even this data lacks proper ID. In other words, there is no distinction for the treatments like Defoliation or De_graining. So, I will make a slight modification to the code.
new_data5= bind_rows (list(DF= Defoliation, DG= De_graining),.id="Treatment")
new_data5
Treatment Genotype Block yield
1 DF CV1 I 50
2 DF CV2 I 52
3 DF CV3 I 55
4 DF CV4 I 56
5 DF CV5 I 54
6 DF CV1 II 52
7 DF CV2 II 50
8 DF CV3 II 56
9 DF CV4 II 58
10 DF CV5 II 59
11 DF CV1 III 52
12 DF CV2 III 50
13 DF CV3 III 56
14 DF CV4 III 58
15 DF CV5 III 59
16 DG CV1 I 62
17 DG CV2 I 59
18 DG CV3 I 62
19 DG CV4 I 60
20 DG CV5 I 59
21 DG CV1 II 61
22 DG CV2 II 60
23 DG CV3 II 63
24 DG CV4 II 62
25 DG CV5 II 60
26 DG CV1 III 64
27 DG CV2 III 58
28 DG CV3 III 65
29 DG CV4 III 61
30 DG CV5 III 62So, we set the Defoliation data as a new variable called “DF” and the De_graining data as “DG,” and we’ll assign the ID (column name) for these as “Treatment.”
Although the rearrangement of the data is similar to using the data.frame() function, the time taken has been significantly reduced. Therefore, using a suitable package for rearranging data like this is much more efficient.